
RNA sequencing (RNA-Seq) is the most popular type of next generation sequencing (NGS) approach, enabling researchers to study the transcriptome qualitatively and quantitatively. However, the term “RNA-Seq” encompasses a wide range of NGS assays, which differ in several technical aspects, including:
- Library construction
- Sequencing technology
- Data analysis
Each assay provides different information about the transcriptome; there's no one-size-fits-all solution. Let’s compare the different techniques and then discuss how to select the most appropriate one for your project.
RNA-Seq Techniques
Here's a table comparing several common RNA-Seq assays, followed by a short description of each. Gaining familiarity with these techniques will help you make an informed decision when designing your experiment.
Comparison of RNA-Seq assays
| Assay Type |
Target RNA |
RNA Selection Method |
Starting Material |
Relative Cost |
| mRNA-Seq |
mRNA |
Poly(A) selection |
Total RNA, cells, tissues |
$ |
| Total RNA-Seq |
mRNA + lncRNA |
rRNA depletion |
Total RNA, cells, tissues |
$$ |
| Strand-Specific RNA-Seq |
mRNA and/or lncRNA |
Poly(A) selection or rRNA depletion |
Total RNA, cells, tissues |
$$ |
| Small RNA-Seq |
Small non-coding RNAs (miRNA, siRNA, piRNA) |
Size fractionation (with adapter ligation to 5' phosphate) |
Total RNA, cells, tissues |
$$ |
| Ultra-Low Input RNA-Seq |
mRNA |
Poly(A) selection |
Total RNA (<500 ng) or cells (<10,000) |
$$$ |
| Single-Cell RNA-Seq |
mRNA |
Poly(A) selection after fractionation of individual cells |
Cells |
$$$$ |
| Iso-Seq |
mRNA |
Poly(A) selection |
Total RNA |
$$$$ |
mRNA = messenger RNA; lncRNA = long non-coding RNA; miRNA = microRNA; siRNA = small interfering RNA; piRNA = Piwi-interacting RNA; rRNA = ribosomal RNA
Messenger RNA-Seq (mRNA-Seq)
In eukaryotes, mRNA transcripts contain polyadenylated tails, which are used to enrich these molecules through poly(A) selection. In this process, total RNA is isolated and subjected to either hybridization with oligo(dT)-conjugated beads/columns or reverse transcription with oligo(dT) primers. Polyadenylated RNA molecules make up just 1-5% of total RNA in many species, meaning sample concentrations after poly(A) selection are typically reduced by a factor of 20-100x. Purified mRNA is converted into cDNA libraries and amplified via PCR to increase library concentration. Poly(A) selection is the most common and the most cost-effective option for eukaryotic mRNA library preparation.
Total RNA-Seq
Total RNA-Seq is used for comprehensive analyses of protein-coding and long non-coding RNA (lncRNA). The latter has important regulatory functions in the genome and is of interest to molecular biologists due to their capacity for epigenetic regulation of transcriptional activity. Since many lack a poly(A) tail, lncRNA molecules are often excluded from poly(A) selection. In total RNA-Seq, oligos complementary to single-stranded rRNAs are used to capture and deplete these molecules prior to sequencing.
Strand-Specific RNA-Seq
Transcript polarity is important for correct annotation of genes. Since there are many genomic regions that generate transcripts from both strands, identifying the polarity of a given transcript provides essential information about the possible function of a gene. However, the polarity of transcripts can be lost during cDNA synthesis and subsequent amplification. Strand-specific RNA-Seq, also known as ‘stranded’ or ‘directional’ RNA-Seq, preserves this information during library preparation, allowing researchers to determine the orientation of the gene on the DNA template. It can be used in conjunction with mRNA-Seq and total RNA-Seq. Multiple methods for creating stranded RNA-Seq libraries are available, such as labeling the second-strand cDNA with uracil (see figure below). Learn more about stranded versus non-stranded RNA-Seq.
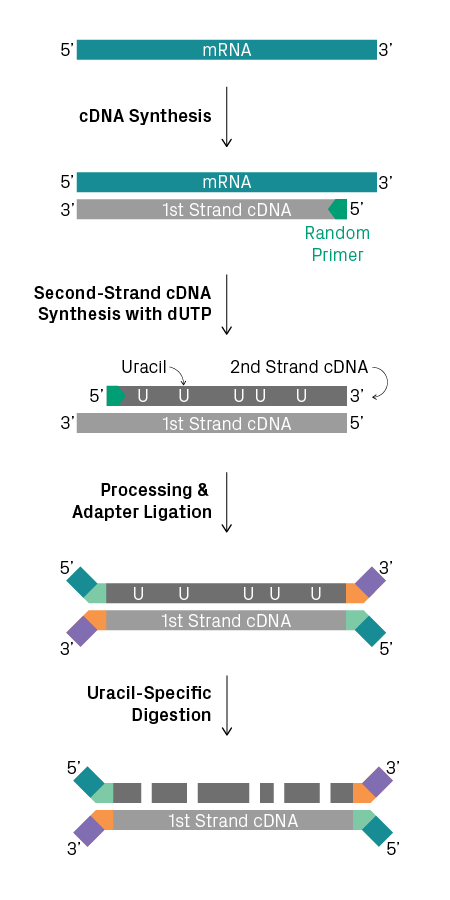
Strand-specific RNA library preparation. Incorporation of dUTP during second-strand synthesis and subsequent uracil-specific digestion selects for the first-strand cDNA.
Small RNA-Seq
Short RNA transcripts, such as microRNA (miRNA) and small interfering RNA (siRNA), also play important gene regulatory functions in the cell. In small RNA-Seq, RNA species are selected by size fractionation from total RNA. Library preparation typically includes ligation of sequencing adapters to 5’ phosphate ends, which are found in Dicer-modified small RNAs but absent in degraded fragments of larger RNA molecules, such as mRNA. The RNA fragments are then converted to cDNA libraries prior to sequencing. Since small RNA molecules can be as short as 21 nucleotides in length, sequencing configurations with fewer cycles (e.g., 1x50 bp) can be used.
Ultra-Low Input RNA-Seq
Since faithful characterization of the transcriptome depends largely on the quality and quantity of the input RNA, standard RNA-Seq approaches call for an ample amount (>500 ng) of intact RNA. Samples producing lower yields and degraded RNA typically require additional amplification steps, as well as higher depths of sequencing to boost data output. These samples are prone to transcriptional bias and poor read mapping to exons. Ultra-low input methods have been developed to selectively amplify full-length transcripts with minimal bias, allowing researchers to perform RNA-Seq on samples containing a few cells.
Single-Cell RNA-Seq (scRNA-Seq)
Distinct from traditional “bulk” RNA-Seq methods, scRNA-Seq allows researchers to capture the transcriptome of individual cells and uncover heterogeneous patterns of gene expression in complex cellular populations. Microfluidics or microwells are typically used to isolate single cells before library preparation. These methods preserve cellular information by adding unique barcodes to each transcript during isolation, which can be bioinformatically traced back to the cell of origin. For more information, learn how single-cell sequencing works and what to consider before starting a single-cell sequencing project.
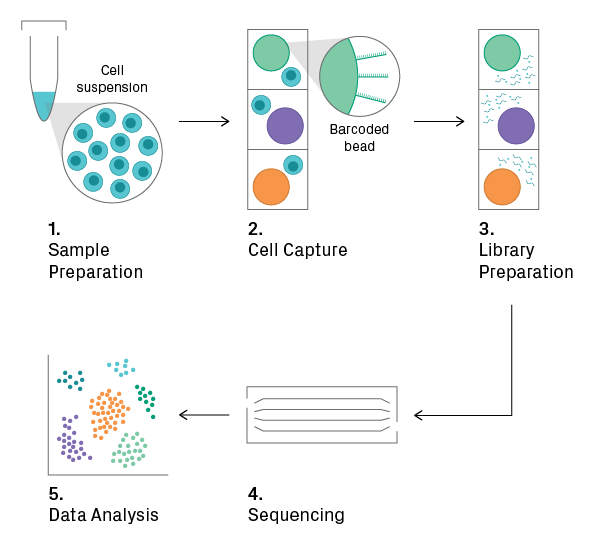
General workflow for scRNA-Seq. (1) The input is a suspension of single cells. (2) The sample is loaded onto a microfluidic chip or cartridge, where cells are partitioned into nanoliter-scale chambers, such as droplets or wells. A unique barcode sequence is added to each chamber, using oligonucleotide-tagged beads, for example. (3) Cells are lysed, and cDNA library construction begins. Within the microchamber, the cellular barcode is added to the target sequences. (4) The pooled library is sequenced. (5) The sequencing data is parsed according to the cell-specific barcodes and analyzed.
Isoform Sequencing (Iso-Seq)
Alternative splicing results in multiple isoforms being encoded by a single gene, which can be effectively analyzed by Iso-Seq. Developed by PacBio®, Iso-Seq uses long-read technology to sequence transcripts contiguously from end to end, eliminating the need for reconstruction. The result is unambiguous information about a transcript’s start, polyadenylation, and splice sites from a single read. Iso-Seq characterizes the full complement of isoforms across the transcriptome, with potential applications including better-annotated genomes, detection of gene fusions, and discovery of novel isoforms.
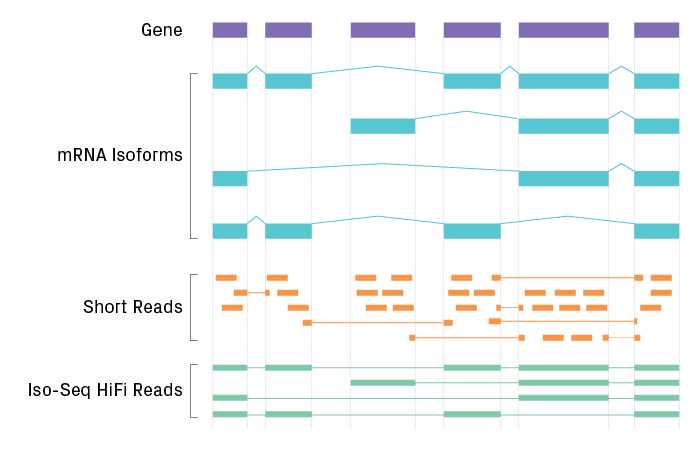
Comparison of RNA sequencing by short-read technologies and Iso-Seq. Short-read technologies enable deeper sequencing coverage for quantitative measurement. Iso-Seq captures sequences of full-length RNA with high certainty.
Selection Guide
Choosing the right RNA-Seq assay for your project depends on several factors, including:
- Experimental objectives
- RNA species of interest (coding, non-coding, or both)
- Starting material
- Availability of a reference transcriptome for your organism
The most important consideration is the objective of your experiment. It's imperative to define your biological question precisely. Having a clear and specific objective will help you determine what type of NGS data is needed, and, in turn, which RNA-Seq assay is the most appropriate. Below is a simplified decision tree that can help guide your selection.
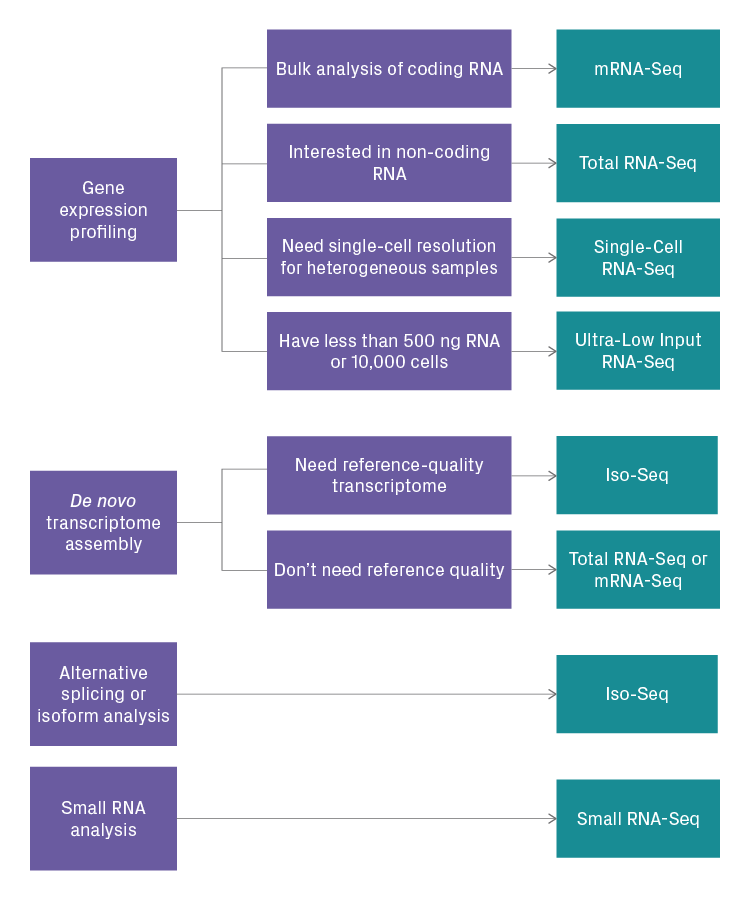
Decision tree for selecting an RNA-Seq assay. The blue boxes indicate the objectives or requirements of the RNA-Seq study, and the blue boxes represent the recommended RNA-Seq assay.
Key Takeaways
- Many types of RNA-Seq are available, each providing different information about the transcriptome
- Having a solid understanding of RNA-Seq and its various forms will help you make an informed decision
- Defining your experimental objective is critical to understanding which RNA-Seq assay is most appropriate for your project; the more specific, the better
Conclusion
With so many RNA-Seq options available today, it can be difficult to know which specific assay suits your project best. By considering a few factors, you can quickly zero in on the right answer. The guidelines in this article are by no means exhaustive, but they provide a good starting point when designing your NGS project.
For a deeper dive into RNA-Seq, download our eBook, A Guide to RNA-Seq. It covers the what, why, and how of this popular NGS technique and offers valuable information to help you achieve the best results for your RNA-Seq experiment.
Download eBook