Thinking about leveraging single-cell sequencing for your research? You’re not alone. Named Method of the Year in 2013 by Nature1, single-cell sequencing has since risen in popularity and expanded its applications over the past decade2, making analysis of transcriptomes, epigenomes, and immune repertoires at cellular resolution possible. Based on next generation sequencing (NGS), many variations of the technology exist, including:
- Single-cell RNA-Seq
- Single-cell ATAC-Seq (assay for transposase-accessible chromatin by sequencing)
- Single-cell immune profiling
- Single-cell multiomics (i.e., RNA-Seq + ATAC-Seq)
With several commercial platforms available, high-throughput single-cell sequencing is now more accessible than ever. In this article, we’ll discuss the three most important factors you’ll need to consider before greenlighting your single-cell sequencing project.
1. Project Goal
Before deciding to use any tool, determining your objective is paramount. Single-cell sequencing is all about uncovering sample heterogeneity, therefore your research objective must align with this purpose. Are you trying to accomplish any of the following?
- Characterizing cell types in a complex sample
- Analyzing the diversity of cellular subgroups
- Discovering rare subpopulations
- Confirming that a sample (e.g., cell line) is homogeneous
If yes, then single-cell sequencing may be appropriate. For situations where you’re solely interested in the aggregate expression profile for a subset of cells, consider using fluorescence activated cell sorting (FACS) or magnetic cell separation to isolate the subpopulation. This assumes that the cells of interest contain a surface marker and an appropriate antibody is available. The isolated cells can then be analyzed by ultra-low input bulk RNA sequencing.
You should only move forward with a single-cell method if traditional bulk sequencing—where the results of thousands of cells are averaged—will not provide the data you need. Bulk RNA and ATAC sequencing are less expensive and simpler to execute.
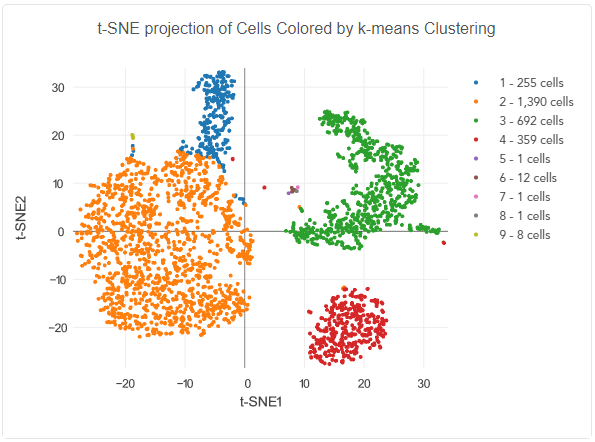
Example data for single-cell RNA sequencing. A t-SNE projection can visualize high-dimensional data, making it easier to identify subgroups of cells, represented as clusters in the plot.
2. Sample Type
The input for single-cell sequencing is a suspension of cells or nuclei; whole tissue or cell clumps cannot be used directly. For library preparation, it’s necessary to physically manipulate single cells or nuclei into individual compartments, such as droplets (see figure below). Let’s examine whether your sample type is acceptable for single-cell sequencing by considering three parameters: feasibility, quality, and quantity.
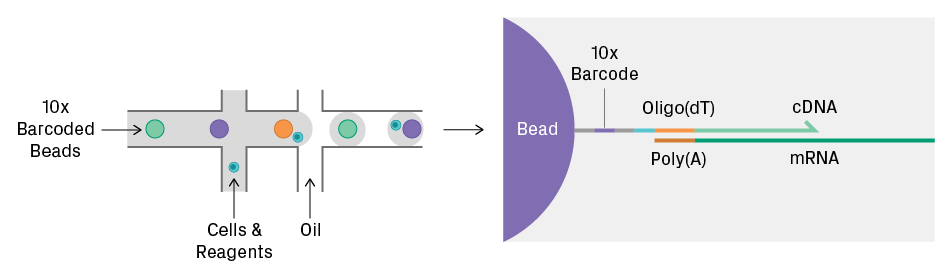
Cell partitioning and library preparation via the 10x Genomics® Chromium™ platform. On a microfluidic chip, cells are encapsulated within droplets containing barcoded gel beads and reagents. To ensure no more than one cell per droplet, the input cell suspension must be free of clumps and diluted to the manufacturer’s specifications. Following cell lysis, the beads capture the polyadenylated mRNA molecules. Reverse transcription generates cDNA tagged with a 10x barcode to identify the cell and a unique molecular identifier (UMI) to label the mRNA transcript.
Feasibility
First, ask yourself: is it possible to generate a cell suspension of your sample? Tissue dissociation can be tricky depending on the sample and tissue type, as the presence of an extracellular matrix can hinder the release of individual cells. Even if possible, the dissociation process may damage or otherwise perturb cells so the resulting data is not biologically meaningful. Check the literature to see if relevant dissociation protocols are available or if others have shown successful dissociation of your sample type.
An alternative approach is to directly isolate intact nuclei, which can be much easier for complex tissues and organs, such as those in the central nervous system3. Notably, nuclei can be isolated from frozen or fixed tissue, adding convenience to sample prep. For single-cell RNA sequencing, keep in mind that observed gene expression may differ between whole cells, which include the cytoplasm, and nuclei alone4.
Quality
When using live cells, a high fraction must be live and intact, otherwise data quality will suffer, meaning lower useable reads and more background noise. For cells, at least 70% should be viable, but greater than 90% is recommended. For samples with low viability, however, dead cell removal can be performed. This procedure uses magnetic beads conjugated to antibodies against annexin V, a marker for dead and apoptotic cells. The beads capture dead or dying cells, enriching the sample for live cells (see figure below). If you’re concerned your sample won’t be able to achieve sufficient viability after dissociation, you can bypass this approach entirely with direct nuclear isolation.
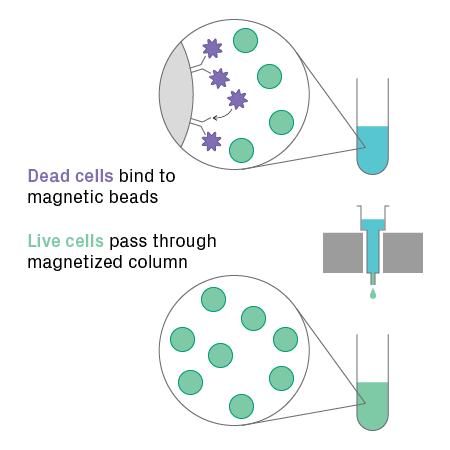
Dead cell removal process. Magnetic bead-conjugated antibodies, which recognize apoptotic and necrotic cell surface markers, are added to a cell suspension. Dead and dying cells bind to the beads and are trapped by a magnetized column, allowing live cells to pass through.
Quantity
Last, the suspension must have enough cells or nuclei. Input requirements will depend on the specifics of your project (single-cell platform, target number of cells, etc.), but at least 50,000 cells/nuclei are required. For best results, aim for 1,000,000, especially if performing dead cell removal.
3. Budget
Not surprisingly, single-cell techniques are generally more expensive than bulk NGS strategies—often by a significant amount. The difference can easily be 10 times or more on a per-base-of-data basis, with extra hardware, reagents, consumables, and labor adding to the cost. However, single-cell sequencing has the potential to provide a trove of data and insights, unobtainable by traditional NGS techniques, justifying the higher price tag.
The cost for a single-cell experiment depends greatly on the details of the project, such as the number of cells targeted and reads per cell. These parameters can typically be adjusted to match your project goals and budget.
Key Takeaways
- The goal of single-cell sequencing is to analyze the heterogeneity of a sample—your project objective must be the same
- You’ll need to generate a high-quality cell/nuclei suspension with at least 50K cells or nuclei
- Providing an unparalleled view of complex samples, single-cell sequencing comes at a justifiably higher price than traditional NGS
Conclusion
Single-cell sequencing methods are incredibly powerful tools for researchers looking to analyze complex cellular populations. They provide high-resolution views of the transcriptome, epigenome, and immune repertoire that are not possible through conventional bulk sequencing. Though at a higher price point for NGS, single-cell approaches can offer unparalleled data that drives novel discoveries.
The guidelines in this article are by no means exhaustive but provide a great starting point as you consider single-cell sequencing for your research. However, it’s important to remember that every NGS project is different and has its own considerations. If you still have questions on whether single-cell sequencing is the best approach for you, feel free to reach out to one of our technical experts and we’d be happy to discuss your project.
Ask an Expert
References
1. Method of the Year 2013. Nat Methods 11, 1–1 (2013).
2. Tang, X., Huang, Y., Lei, J., Luo, H. & Zhu, X. The single-cell sequencing: new developments and medical applications. Cell Biosci 9, (2019).
3. Grindberg R. V et al. RNA-sequencing from single nuclei. Proc. Natl Acad. Sci. USA 110, 19802–19807 (2013).
4. Bakken, T. E. et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE 13, e0209648 (2018).