Azenta Life Sciences and PacBio® held a virtual symposium titled Decoding the Complexity of Human Health – A HiFi Vision on October 5, 2021 that explored applications of high-fidelity (HiFi) long-read sequencing within human biomedical research. Speakers from top academic research labs and biotech shared their cutting-edge work with this next generation sequencing (NGS) technology. Here, we recap the six presentations and discuss the themes that emerged from the event.
1. PacBio Long-Read Sequencing for Multiplexed Joint Epigenetic and Genetic Profiling
Presented by Michael Kladde, Ph.D., Professor of Biochemistry and Molecular Biology, University of Florida
Challenge: Mid-scale methods to simultaneously profile DNA methylation and chromatin accessibility are limited and expensive
Genetic and epigenetic analysis based on NGS can be performed at various scales: at the low end, multiplex PCR followed by NGS can target a few loci; at the high end is whole genome sequencing. Mid-scale methods that capture dozens to hundreds of targets often rely on a Capture-Seq approach using RNA bait hybridization. Synthesizing these probes—that is, a large set of custom modified oligos—can be costly. Furthermore, short-read sequencing offers limited windows (<300 bp) to explore the interplay between methylation and chromatin accessibility at the resolution of individual DNA molecules.
Solution: MAPit-FENGC enables cost-effective epigenetic analysis of ~150 user-defined loci
A new approach combines four technologies to simultaneously characterize endogenous DNA methylation and chromatin accessibility in about 150 targeted regions with minimal use of expensive modified oligos:
- Methyltransferase Accessibility Protocol for individual templates (MAPit), also known as Nucleosome Occupancy and Methylome Sequencing (NOMe-Seq), labels nucleosome-free regions of genomic DNA with GpC methyltransferase1.
- Flap-Enabled Next-Generation Capture (FENGC), a novel method for selection and excision of genomic DNA, uses standard oligos for targeting; all modified oligos are universal.
- PacBio HiFi sequencing provides full-length reads of the target regions.
- Methylscaper software analyzes the data and provides a framework for visualization2.
The figure below summarizes the workflow. Results show approximately 80% on-target sequencing for regions of 940 bp. High sequencing coverages enable identification of rare epigenetic states (1 in 1000 epiallele frequency) and mechanistic insights. The longer reads reveal more epigenetic features at a distance; for example, the sliding of +1 nucleosomes on a bidirectional promoter.
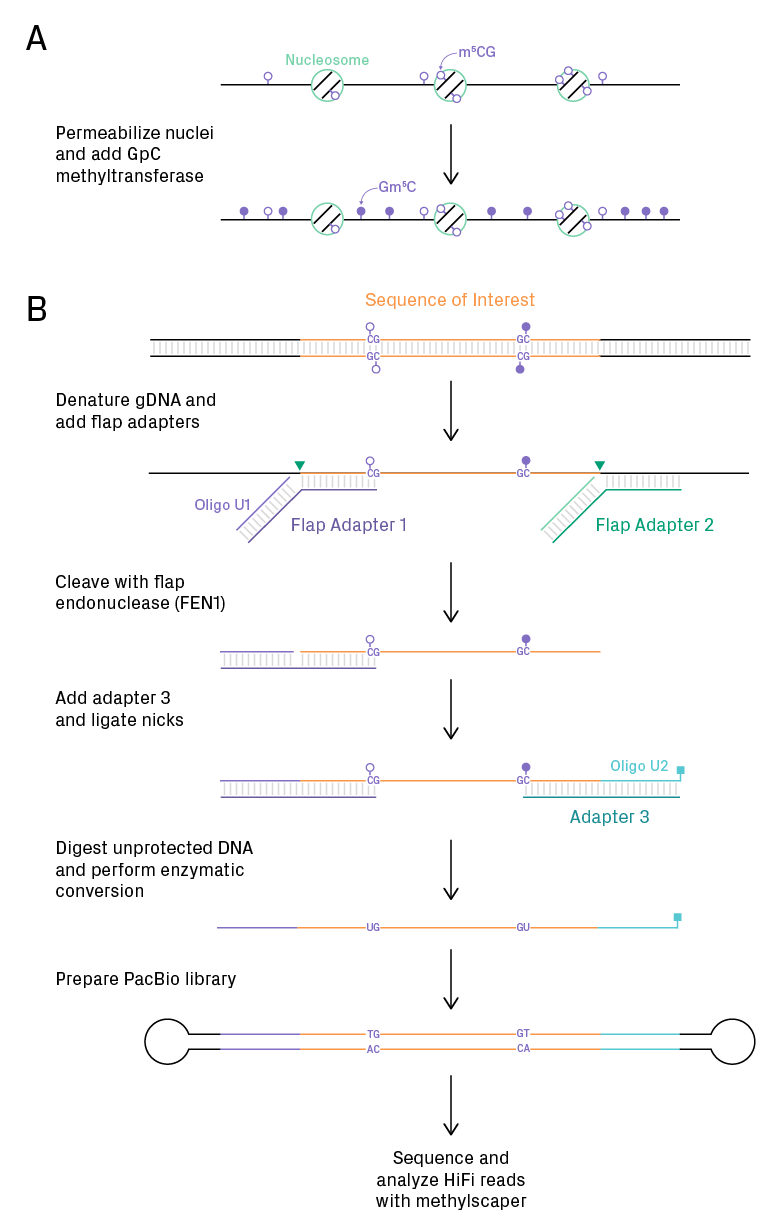
MAPit-FENGC approach with HiFi sequencing. (A) The MAPit method uses digitonin to permeabilize nuclei and GpC methyltransferase to methylate DNA (specifically C in 5’-GC-3’) in protein-free regions. DNA is then rapidly isolated. The resulting DNA retains endogenous methylation (m5CG) in addition to in vitro methylation (Gm5C) for chromatin accessibility. (B) In FENGC, genomic DNA is denatured. Two flag adapters anneal to the template at the ends of the region of interest. Flap endonuclease (FEN1) cleaves at the 5’ of the flap overhangs (green triangles). A third adapter, containing an oligo with a blocking group, anneals to the 3’ end of the fragment. Ligase seals the nicks in the top strand. Exonuclease digestion then removes untargeted DNA. Methylated cytosine is converted to uracil using an enzyme-based method. PacBio libraries are prepared, and the circularized DNA is sequenced repeatedly to generate highly accurate consensus reads. The resulting data is analyzed with methylscaper software. Figure adapted from Dr. Michael Kladde’s slides.
2. High-Resolution StrainID Fingerprinting of Bacteria in Premature Infant Fecal Samples
Presented by Mark Driscoll, Ph.D., Chief Scientific Officer, Shoreline Biome
Challenge: Differentiating pathogenic strains for clinical use typically requires high-coverage whole genome sequencing
Traditional amplicon-based approaches for bacterial identification, such as the V1-V9 regions of the 16S rRNA gene, are insufficient to distinguish strains. Shotgun whole genome sequencing has the potential for strain differentiation but only at high coverages, leading to greater cost and analytical complexity.
Solution: StrainID uses HiFi amplicon sequencing for highly accurate identification of bacterial strains
The StrainID method uses a 2.5 kb amplicon that encompasses the entire 16S gene, part of the 23S gene, and the internal transcribed spacer (ITS) that intervenes (see figure below). Many bacteria have multiple copies of the 16S-ITS-23S locus in their genomes; for example, E. coli has seven and Klebsiella pneumoniae has eight. HiFi sequencing of the amplicon sequence variants (ASVs) enables a genetic fingerprint of the strain. Requiring 200X to 2000X less data per sample than high-coverage shotgun metagenomics, StrainID has sufficient sensitivity and resolution to identify novel bacteria and track them over time in infant fecal microbiomes3.
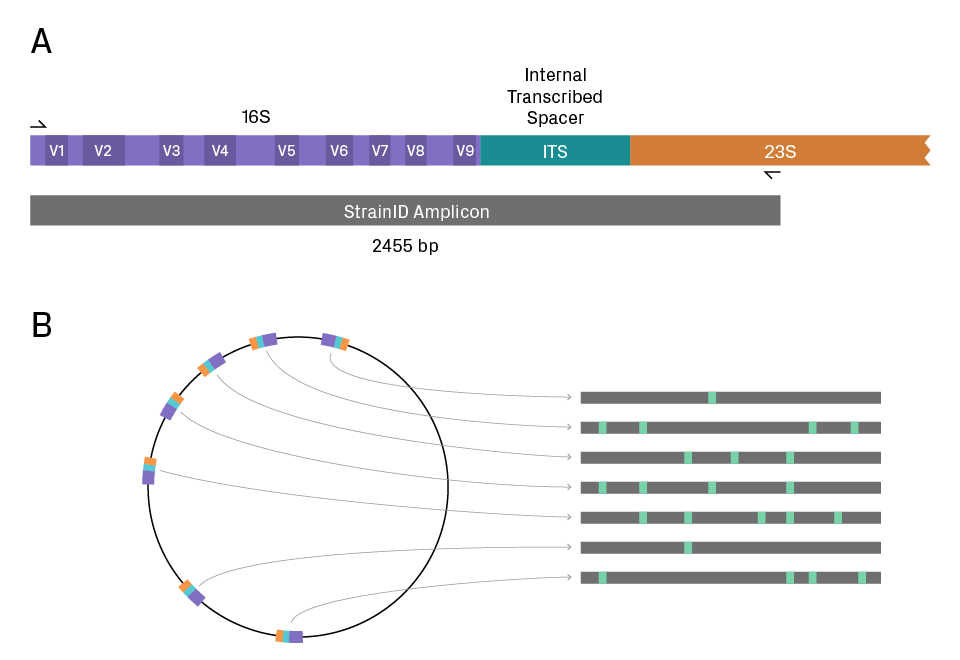
StrainID approach to metagenomics. (A) The StrainID amplicon covers the full 16S gene, ITS, and partial 23S gene. (B) Many bacterial genomes have multiple copies of 16S-ITS-23S, with possible variation between the loci. Taken together, the sequences generate a genetic fingerprint with sufficient resolution to distinguish closely related strains. PacBio HiFi sequencing provides full-length, highly accurate reads of the amplicons and thus high confidence in strain differentiation. Figure adapted from Dr. Mark Driscoll’s slides.
3. Jasmine: Population-Scale Structural Variant Comparison and Analysis
Presented by Melanie Kirsche, Ph.D. Candidate in Michael Schatz's Lab, Johns Hopkins University
Challenge: Standard analysis pipelines significantly overestimate the number of structural variants
Structural variants (SV) are large-scale genomic mutations that come in several varieties, including insertions, deletions, duplications, inversions, and translocations. Trio datasets are often analyzed to find de novo variants, which involves sequencing and comparing the genomes of a child and its parents. Structural variants are considerably trickier to identify than single nucleotide variants. Sequencing and mapping errors complicate the process of determining whether two or more SVs can be called the same. False positives are a major problem with standard variant calling programs, which can overestimate de novo SVs by as much as 200-fold.
Solution: Jasmine, a new software pipeline, provides robust structural variant calling
Jasmine tackles SV calling by representing the variants as points in 2D space based on their chromosomal position and length (see figure below). An algorithm determines if nearby points on the graph should be grouped together, thus representing the same structural variant. Jasmine outperforms widely used comparison methods, demonstrating more than a 5-fold decrease in Mendelian discordance in trio datasets4. It achieves the highest confidence in SV calling with PacBio HiFi reads, as this sequencing data is best suited for capturing structural variants (see next section).

Overview of Jasmine structural variant calling. Initially, per-sample structural variant call sets are grouped by type and reference sequence. The example shows deletions within a region of chromosome 1. The variants are plotted based on chromosomal position and length. Nearby points are connected by edges. A modified version of Kruskal’s algorithm for minimum spanning trees finds the pattern with the smallest sum of edge distances. The resulting groups of connected points are considered unique variants. Figure adapted from Melanie Kirsche’s slides.
4. Increasing Solve Rates in Rare and Mendelian Disease Research with Long-Read Sequencing
Presented by Edd Lee, Director of Marketing, Rare and Inherited Disease, PacBio
Challenge: Many rare and inherited diseases cannot be explained with short-read sequencing methods
Rare diseases, in aggregate, affect 10% of the population, yet more than half of cases remain unsolved after short-read exome or whole genome sequencing. The issue is particularly acute for diseases caused by structural variants (SVs). Large insertions, deletions, duplications, and translocations can be very difficult to identify using short reads. Several medically relevant genes lie in “dark” genomic regions that cannot be assembled or aligned using short-read sequencing methods.
Solution: PacBio HiFi reads provide comprehensive variant detection, especially for structural variants
PacBio sequencing technology produces highly accurate long reads up to 15 kb with greater than 99.9% accuracy. It has the best performance for detecting all variant classes, including single nucleotide variants (SNVs), indels, and structural variants (see figure below). In the 2020 precisionFDA challenge, PacBio HiFi reads outperformed both short reads and noisy long reads5. Of 193 medically relevant genes with coverage issues (i.e., those located in dark regions), PacBio sequencing can detect 152 with 100% coverage. Thus, this technology has the power to elucidate the genetics behind many rare and heritable diseases.
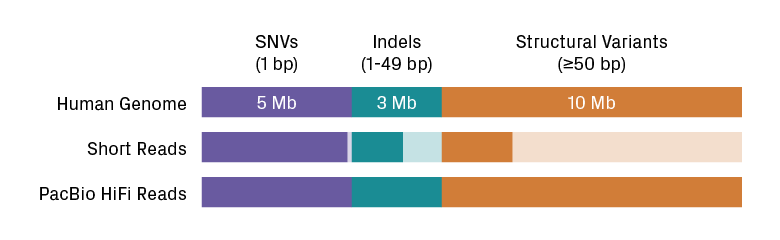
Variant detection by sequencing technology. Although fewest in number, SVs take up the most genomic space in the human genome. PacBio HiFi reads outperform short reads for all variant types, especially SVs. Figure adapted from Edd Lee’s slides.
5. Single-Molecule Long-Read Sequencing Reveals a Conserved Intact Long RNA Profile in Sperm
Presented by Xin Li, Ph.D., Associate Professor, University of Rochester Medical Center
Challenge: Short-read sequencing is inadequate for profiling long transcripts in sperm
Sperm were considered a bottleneck for the transmission of epigenetic information across generations. The cells lose most of their cytosol and histones during development, and methylation patterns from paternal DNA are erased in early embryogenesis. However, miRNA from sperm was recently discovered to convey transgenerational effects. Are there mRNA transcripts in sperm that can do the same? Answering this question requires first identifying whether sperm cells contain intact mRNA. Previous work suggested that mRNA in sperm is highly fragmented. Short-read sequencing suffers from the inability to distinguish between fragmented and intact mRNA, as RNA fragmentation is an early step in NGS library preparation.
Solution: Iso-Seq uses long-read technology to sequence transcripts contiguously from end to end
Sequencing on the PacBio platform enables full-length HiFi reads of transcripts. This approach, called isoform sequencing (Iso-Seq), can therefore distinguish between fragmented and long intact mRNA. It also provides unambiguous information about the transcript’s start, polyadenylation, and splice sites from a single read (see figure below). Analysis of the sperm transcriptome with Iso-Seq identified 3,440 long intact RNA species, of which 2,479 were novel isoforms and 198 were novel loci6.
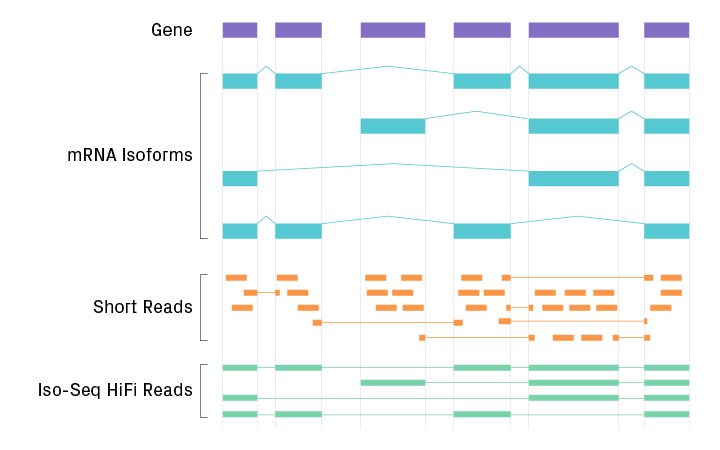
Comparison of RNA sequencing by short-read technologies and Iso-Seq. HiFi reads can capture the full-length RNA sequence with high certainty.
6. Application of PacBio Long-Read Sequencing in Oncology AAV-Mediated Gene Therapy Development
Presented by Elizabeth Louie, Ph.D., Supervisor, Technical Applications, Azenta Life Sciences
Challenge: Targeted AAV therapy success is reliant upon the quality and accuracy of the packaged virus
Adeno-associated virus (AAV) is gaining popularity as a gene therapy vector for oncology applications. A recombinant AAV (rAAV) genome carries a transgene of up to ~4.5 kb, flanked by two inverted terminal repeat (ITR) regions. Contaminant DNA or mutated rAAV sequences introduced during viral packaging can affect clinical safety and efficacy. Thus, it’s imperative to examine genome integrity and measure the heterogeneity of packaged material, as part of quality control.
Solution: PacBio long-read sequencing thoroughly characterizes packaged AAV DNA
Sequencing on the PacBio platform enables full-length HiFi reads of the rAAV genome, providing comprehensive analysis of mutations in the packaged DNA. It can identify truncation hotspots and recombination events as well as quantify the abundance of each variant (see figure below). ITR regions, which are notoriously unstable, can be sequenced with high accuracy. Azenta Life Sciences has optimized library preparation for the PacBio platform and developed comprehensive and customizable analysis pipelines to assess rAAV integrity, as part of its AAV genome sequencing services.
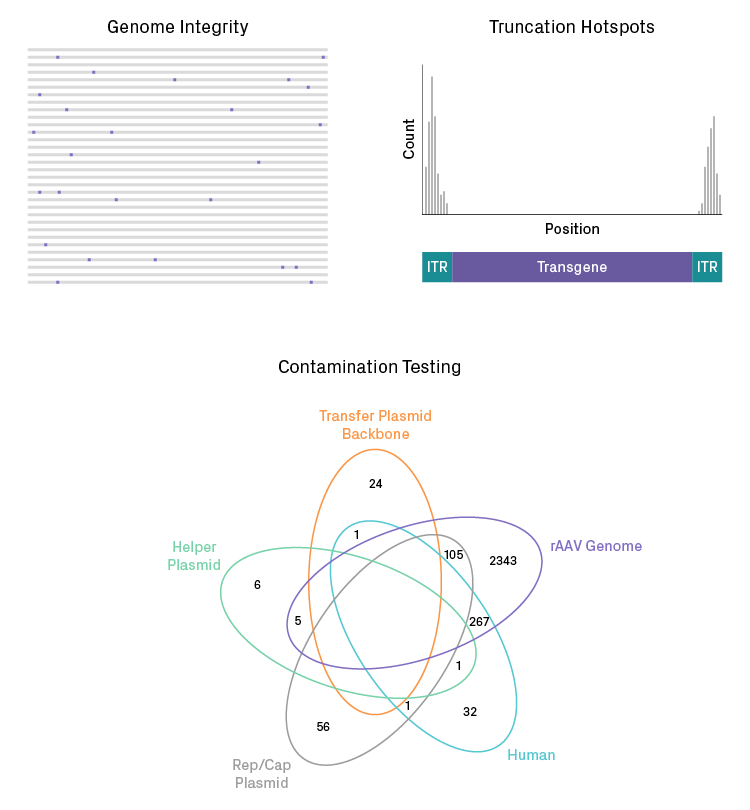
Examples of data analysis for PacBio long-read sequencing of packaged rAAV particles. HiFi reads enable comprehensive analysis of genome integrity and quantitative detection of truncations and contaminant sequences.
Key Takeaways
- The PacBio platform can generate high-fidelity (HiFi) reads up to 15 kb with over 99.9% accuracy
- HiFi sequencing is a powerful tool to explore larger features of the genome, epigenome, or transcriptome at the level of single DNA/RNA molecules
- It has widespread applications to biomedical research, including the investigation of human cells, pathogens, and viral vectors
Conclusion
PacBio sequencing provides highly accurate long reads that are ideal for analyzing larger features of the genome, epigenome, or transcriptome, especially when accurate reconstruction from short reads isn’t feasible. By capturing more information (of high quality) per read, HiFi sequencing enables powerful tools to explore chromatin structure, monitor bacterial pathogens, identify structural variants, discover novel transcripts, and assess the quality of gene therapy products. For more details, watch the full recording of the Decoding the Complexity of Human Health – A HiFi Vision virtual symposium.
Have a question about PacBio HiFi sequencing? Feel free to reach out to one of our technical experts. We’ll gladly discuss your project and help you figure out if long-read sequencing is the best solution.
Ask an Expert
References
1. Kilgore, J., Hoose, S., Gustafson, T., Porter, W. & Kladde, M. Single-molecule and population probing of chromatin structure using DNA methyltransferases. Methods vol. 41 320–332 (2007).
2. Knight, P. et al. Methylscaper: an R/Shiny app for joint visualization of DNA methylation and nucleosome occupancy in single-molecule and single-cell data. Bioinformatics (2021).
3. Graf, J. et al. High-Resolution Differentiation of Enteric Bacteria in Premature Infant Fecal Microbiomes Using a Novel rRNA Amplicon. mBio vol. 12 (2021).
4. Kirsche, M. et al. Jasmine: Population-scale structural variant comparison and analysis. bioRxiv (2021).
5. In precisionFDA Challenge, PacBio HiFi Reads Outperform Both Short Reads and Noisy Long Reads. PacBio Blog (2020).
6. Sun, Y.H., Wang, A., Song, C. et al. Single-molecule long-read sequencing reveals a conserved intact long RNA profile in sperm. Nat Commun 12, 1361 (2021).